Retinitis Pigmentosa
What Is Retinitis Pigmentosa?

Retinitis pigmentosa (RP) is the most common of a large group of progressive retinal degenerations or dystrophies [i.e., degenerative disorders].
There is considerable overlap among the various types. It usually refers to a group of hereditary conditions involving one or several retina layers, causing progressive degeneration.
Some Facts about the Retina
The retina is the light-sensitive tissue that lines the inside surface of the eye.
The retina contains photoreceptor cells that convert (or process) incoming light into electrical impulses. These electrical impulses are carried by the optic nerve (which resembles your television cable) to the brain, which finally interprets them as visual images.
There are two types of photoreceptors: rods and cones, the light-processing cells responsible for peripheral (side) and central (straight-ahead) vision.
Rods:
- The specialized, highly light-sensitive retinal processing cells can function in low light levels. They provide peripheral (or side) vision, are responsible for dark adaptation, and are most sensitive to movement/motion. They are less sensitive to color perception.
- A normal retina contains approximately 120-150 million rods, primarily in the peripheral, or outer, retina.
- Rods provide scotopic vision which refers to eyesight in low-light conditions.
Cones:
- The specialized retinal processing cells function in bright light levels and provide central (or straight-ahead) vision, along with sharp visual acuity, detail, and color vision. They require bright light to function and are not sensitive to lower light levels.
- A normal retina contains approximately 6-7 million cones, primarily in the macula, the small area in the center of the retina that provides clear central vision. Cones are the most concentrated in the fovea, located in the macula’s center and provides the sharpest detail vision.
- Cones provide photopic vision, which refers to eyesight in daylight conditions.
In most retinitis pigmentosa (RP) and RP-like diseases, the rods deteriorate first, followed by the cones.
With RP, central vision is usually retained until late in the disease as the peripheral vision slowly constricts.
What Causes Retinitis Pigmentosa?
Retinitis pigmentosa is an inherited condition caused by mutations in genes important for rod and cone function. While many genes can cause RP, a single gene is responsible for each affected individual or family. Genetic testing can reveal which gene is responsible in a given individual.
Suppose you have been diagnosed with retinitis pigmentosa. In that case, you may wish to undergo genetic testing with the aid of a genetic counselor, who will also ask questions about your family history and family tree (called a “genetic pedigree”). Usually, this is managed by a retinal specialist, who may also enlist the help of “super-specialists.” A super-specialist is a physician with additional, highly specific training in a specialty area of medicine, and this case refers to a retinal dystrophy specialist.
Retinitis pigmentosa occurs in approximately 1 in 4,000 people in the United States. A related condition, Usher syndrome, includes retinitis pigmentosa accompanied by significant hearing loss from birth.
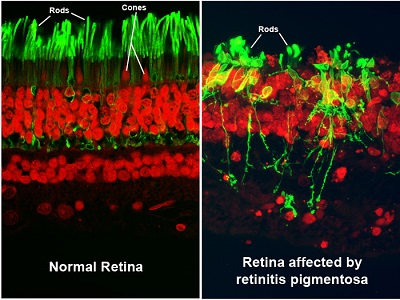
What Are the Symptoms and Signs of Retinitis Pigmentosa?
Since retinitis pigmentosa is a rod dystrophy, and rods are responsible for night vision, you will start to notice an increasing difficulty in night vision as the first symptom, followed by progressive constriction, or closing in, of the visual field [i.e., the total area an individual can see without moving the eyes from side to side]. This may progress to “tunnel vision,” which is considered legal blindness once the visual field is 20 degrees or less in the better-seeing eye. For comparison, a normal visual field is approximately 160 degrees.
Rarely, total blindness may eventually occur. In some younger patients, in addition to the visual findings, poor hearing or deafness may occur. Significant visual loss may not occur until later in life. Some patients retain useful central vision their entire life.
Portions of this article were published originally at www.medicinenet.com/retinitis_pigmentosa/article.htm
By Frank J. Weinstock, MD,
Edited and Updated in 2024 by Abigal Fahim, MD